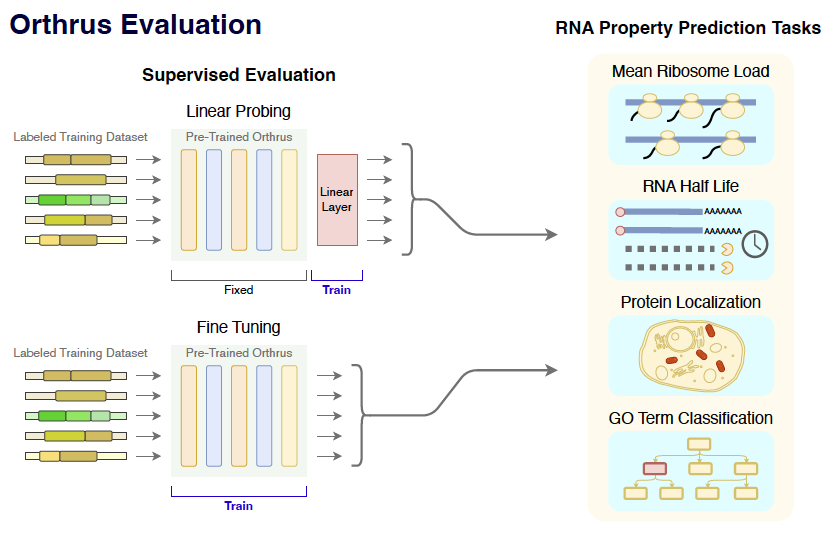
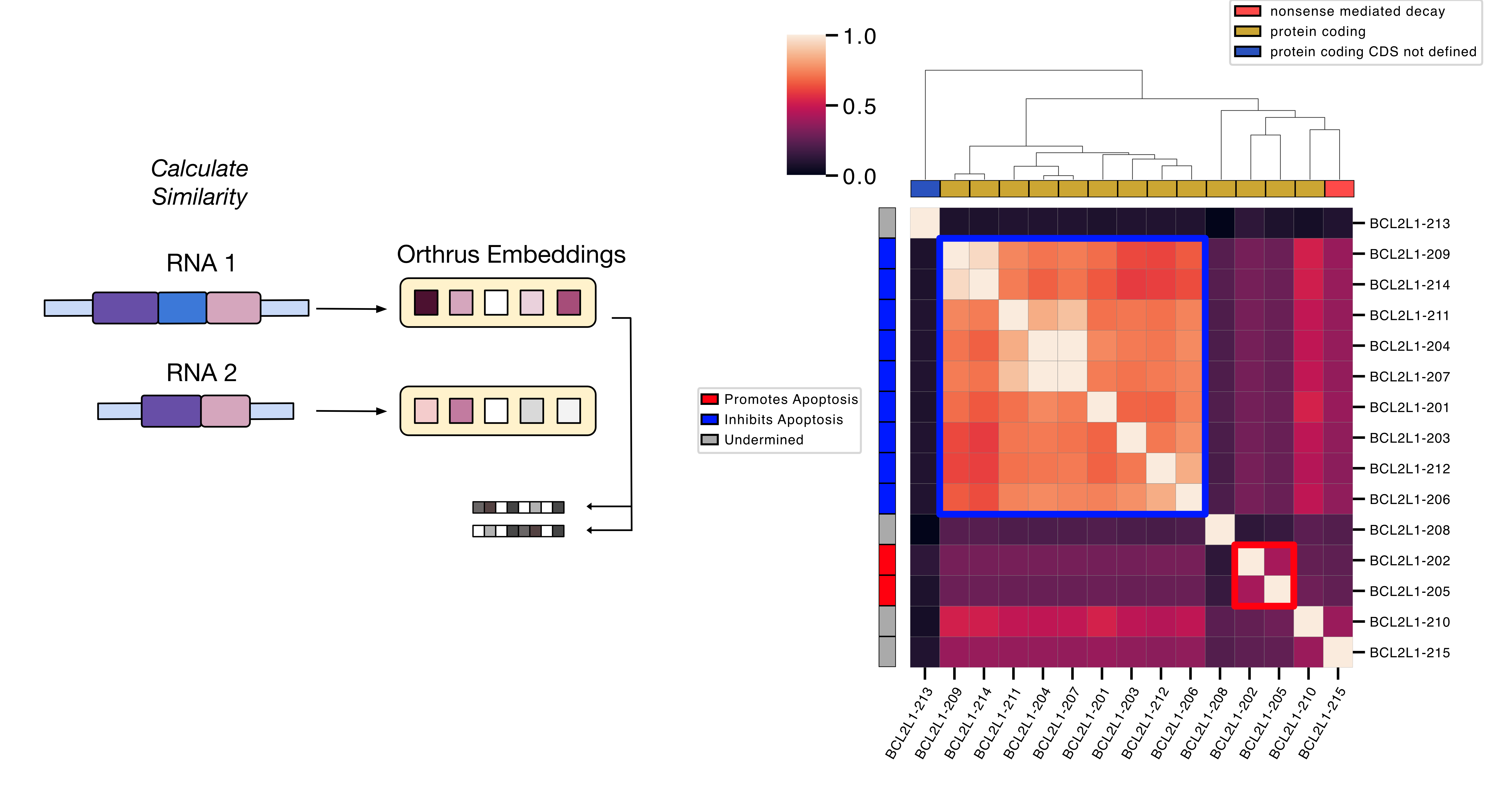
The Orthrus foundation model is trained to accurately represent the properties of mature RNA transcripts in both
regulatory and functional aspects. We evaluate Orthrus and other genomic FMs on the following prediction tasks:
-
RNA Half Life:
This regression task measures the decay rate of mRNA in cells, and
is an important cellular property due to its implications for protein expression regulation.
We use the
Agarwal and Kelley (2022) dataset for this benchmark, which consists of 10,432 human and
11,008 mouse RNA sequences with corresponding measurements.
-
Mean Ribosome Load:
This task estimates the translational efficiency of
a given mRNA molecule, measuring the number of ribosomes translating a single
mRNA molecule at a point in time. Accurate MRL measurement offers
insights into the efficiency of protein translation, a key process in cellular function.
The dataset in question,
derived from the HP5 workflow, captures this metric across 12,459 mRNA isoforms from 7,815 genes.
-
Protein Subcellular Localization:
Protein function is often linked to its subcellular location, which can be determined
using cells that are immunofluorescently stained. We downloaded a dataset of 10,409 genes,
whose protein localization was determined by the
Human Protein Atlas, and benchmark the ability to classify
between the 12 most common localizations for the canonical isoform per gene, as determined by
Appris database.
-
Protein GO Term Classification:
Gene ontology (GO) terms are a hierarchical classification
system used for assigning function to genes and their products. GO term hierarchical systems
allow for fine-grained annotation of function, with broader terms at the top of the
hierarchy and increased specificity closer to the bottom. To annotate genes with gene
ontology terms, we subset GO classes three levels from the root, labeling all available
genes.
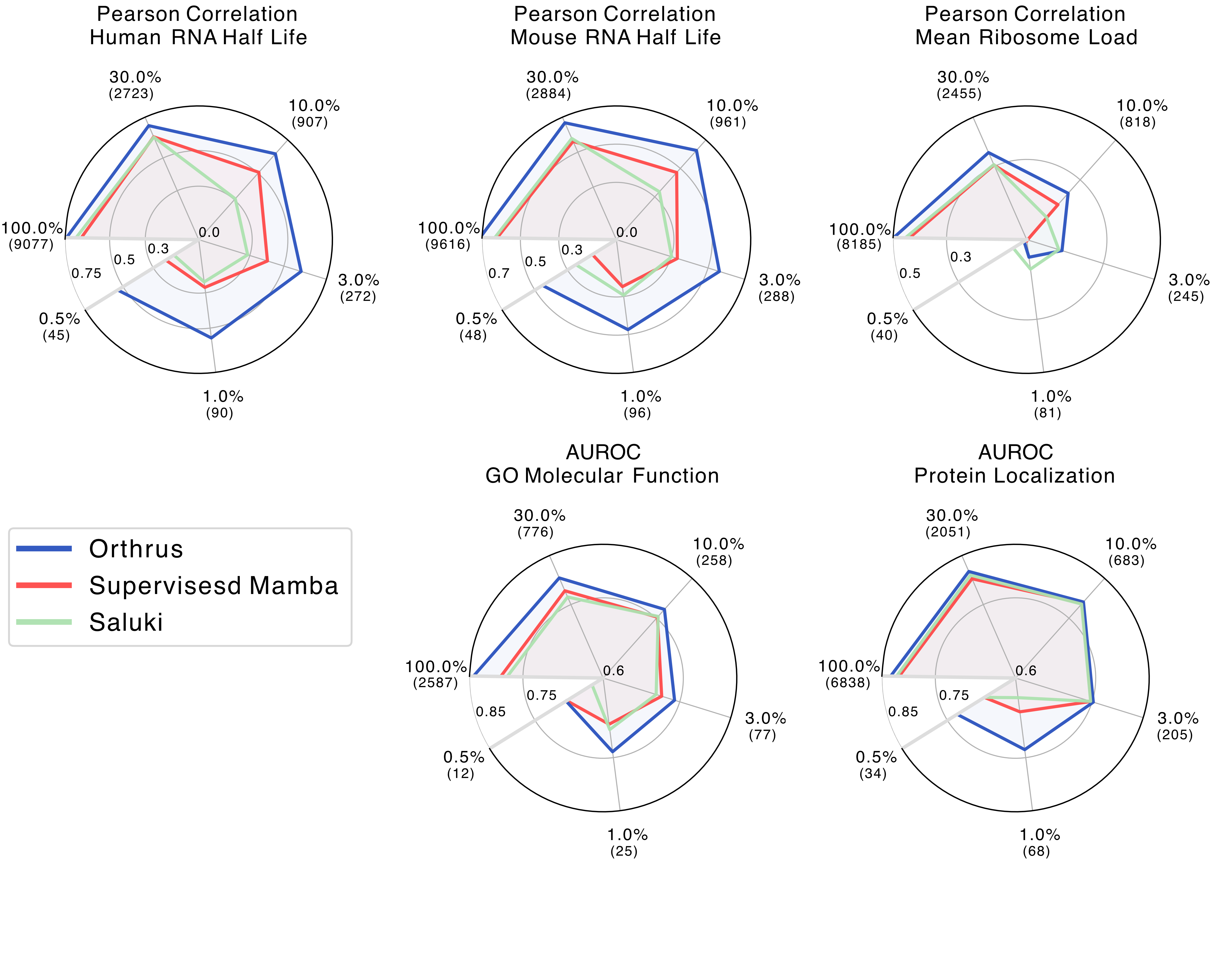
We benchmark the performance of Orthrus against several genomic foundation models, as well as supervised
task-specific models in both linear probing and fine tuning contexts. For linear probing, we simply train
a linear model on top of fixed FM embeddings, while for fine-tuning we perform end-to-end training on the
labelled prediction dataset. Overall, we see that Orthrus outperforms existing genomic FMs, while approaching
the performance of supervised models designed specifically for RNA property prediction such as
Saluki.
In our experiments, by just fitting a linear model on trained Orthrus embeddings, Orthrus is able to achieve tuned DNN performance.
Orthrus also shows strong data efficiency for fine-tuning,
highlighting its potential to be used for extrapolating small experimental datasets.
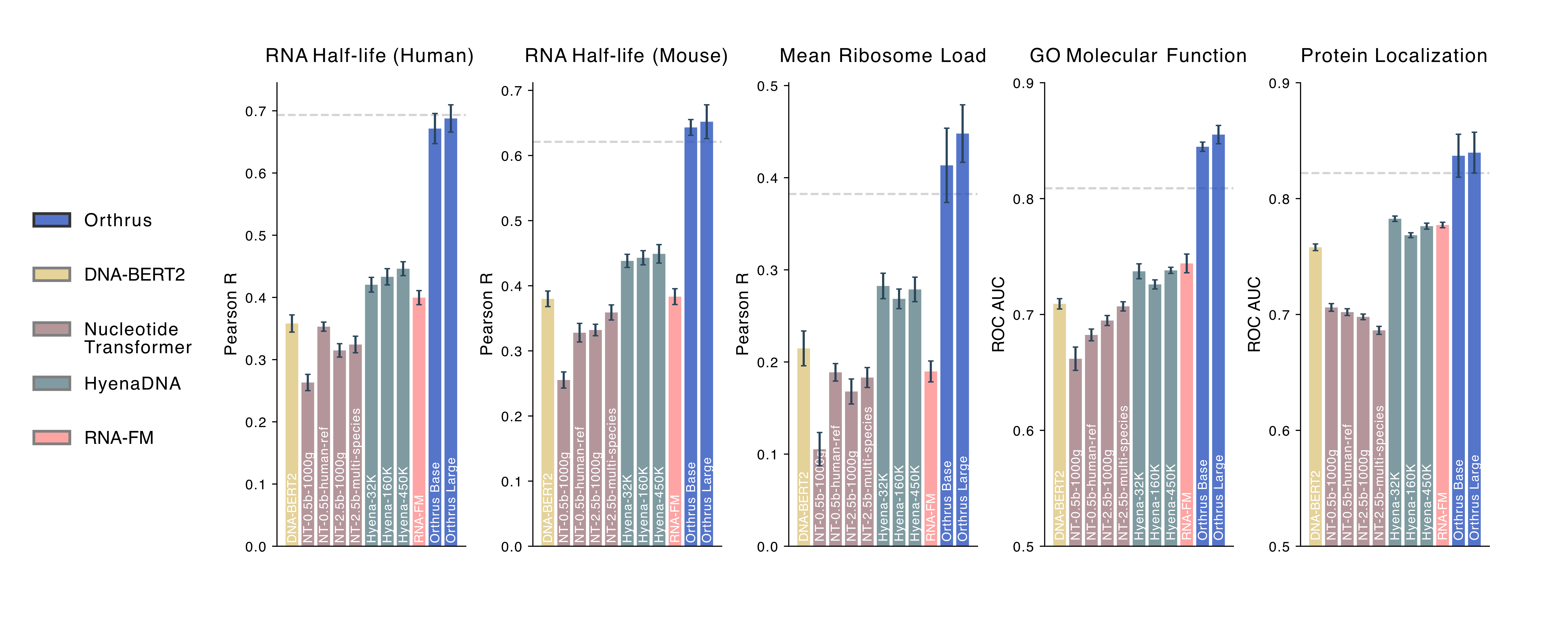
Benchmarking linear probing performance on RNA property prediction tasks for self-supervised genomic
foundation models. Individual bars represent the performance of foundation model variants, which
typically differ in parameter count and pre-training dataset. Error bars show 95% confidence intervals,
constructed using 10 runs with randomized data splits. The grey dashed line indicates the performance of the fully
supervised Saluki method trained with access to labels.